1. 마이크로바이옴(Microbiome)의 정의
1.1. Microbiome = Microbe (미생물) + biome (생태계)
- 마이크로바이옴은 특정 환경 내에 서식하는 미생물 집단과 그들의 유전적 물질 (DNA, RNA, 단백질, metabolite)의 총괄
- 토양, 해수 뿐만 아니라 사람의 피부, 구강, 장 등도 미생물 집단이 서식하고 있음
- 인간의 몸에 서식하는 미생물 집단은 단순 기생이 아니라 숙주(인간)와 쌍방향적 소통을 하면서 숙주의 건강 상태와 깊은 연관이 있다는 것이 최근 밝혀지고 있음
- 이들의 세포 수는 숙주의 전체 체세포보다는 최소한 같거나 그 이상이라고 알려져 있고, 유전자 수는 100배 가량 많다고 알려져 있음 (Microbiome 2,000,000 genes vs. Human 20,000 genes, ref[1])
- 유전자 (=기능을 할 수 있는 DNA 단위)가 더 많다는 것은 그만큼 더 많은 종류의 RNA, 단백질 (효소), 그리고 대사물질을 생성해낼 수 있다는 것을 뜻함
- 숙주의 생리가 조절되는 데 있어서 숙주 유래 물질들 만큼이나 마이크로바이옴 유래 물질이 중요하게 작용할 가능성이 매우 높음을 뜻함
- 실제 다양한 질병상황에서 마이크로바이옴의 변화가 확인되었고, 심지어 마이크로바이옴의 회복이 질병의 치료 방법으로 가능성을 보이는 사례들이 보고됨

2. 마이크로바이옴 연구의 트렌드
2.1. Highthroughput Sequencing (HTS) 기반 마이크로바이옴 분석기술의 발달
2.1.1. 마이크로바이옴의 초기 분석과 그 한계점
- 마이크로바이옴은 대표적인 집단유전체의 일종 (=특정 환경 내에서 서식하는 개체들의 집합)
- 장내 균총과 같은 특정 환경 내 미생물의 분포를 확인하는 작업은 예전부터 시도되어 옴
- 그러나 초기의 분석은 배양 기반의 확인이 위주였고, 이는 실제 장내 환경과 매우 상이하기때문에 많은 수의 미생물에 대한 정보를 확인할 수 없고 분포에 bias가 생겨 정확한 상태를 대변하지 못함
ex) Escherichia coli (대장균) : 혐기 조건의 장 내에서 실제로는 매우 소수의 분포를 가지지만, 배양 기반의 분석에서는 major한 species로 측정이 되어 이름 역시 대장을 대표하는 듯한 느낌을 주는 '대장균'이라고 명명된 예시
2.1.2. HTS의 등장과 16S rRNA sequencing기술
- 마이크로바이옴 내 무수한 미생물들의 종류를 보다 정확히 파악하기 위해서는, 배양과 같은 loss와 bias가 많이 발생할 수 있는 방법이 아니라 환경 내 모든 유전정보를 sequencing하는 것이 보다 정확한 확인이 가능한 방법
- 그러나, 이를 수행할만큼 대량으로 유전정보를 sequencing하고 또 분석할 기술이 없어서 한계가 있어왔음
- 한편, 1990년대에는 human genome project의 시대였음
- 그 중에서 expressed sequence tag (EST)라고 불리는 protein 번역에 대한 정보를 가진 mRNA가 어떤 것이 있는지를 규명하기 위한 과정에서 1992년 Sydney Brenner* [2]에 의해 처음 NGS** 기술의 concept이 제안됨
* Sydney Brenner: Lynx Therapeutics INC. 설립자, 이후 Lynx Therapeutics INC.는 Solexa에 합병, 또 이 Solexa INC. 가 Illumina에 합병됨
** NGS: Next-generation sequencing = Massive parallel sequencing
- 이후 컴퓨팅 파워 향상 등의 발전으로 2004년 처음으로 NGS 기술이 상용화되었고, 현재까지 계속적으로 비약적 발전이 이뤄지고 있는 중
- Human genome은 어쨌든 하나의 homo sapiens의 whole genome을 분석하는게 목표라, DNA뽑아서 절편화시켜서 그 조각들을 sequencing & 다시 assembling하는 과정을 거치면됨
- 그렇다면, 마이크로바이옴은?
- 위에서 계속 언급했듯 수많은 미생물들의 집합체이다보니 human genome처럼 하면 fragment assembly에 error rate가 너무 높아질 위험이 있음
- 즉, 시퀀싱 이전에 최소한의 길이로 미생물들을 구분할 수 있을만한 marker gene이 필요했고, 그래서 등장한게 16S rRNA gene의 sequencing!
- 16S rRNA gene은 prokaryotic ribosome의 30S small subunit의 일부분인데, conserved region 사이사이에 약 9개의 variable region (V1~V9)이 섞여 있는 구조 (그림 2, [3])
- 즉, conserved region으로 prokaryotic(= bacteria)인지 판별이 가능함과 동시에 variable region은 동일 species내에서는 차이가 없지만 species간에는 차이가 큰 특징이 있어 생물 동정에 유용하게 사용이 가능
- 보통 conserved region에 annealing이 되고 amplicon내에 variable region이 포함되도록 primer를 design하는 방식
- 16S rRNA 전체를 하기보다는 동정은 가능하면서 보다 효율적인 시퀀싱을 위해 일부 variable region만 포함되게 하는데 대표적으로 V4 region을 사용하곤 함 (효율적인 시퀀싱이 필요한 이유는 가격 측면도 있지만, 특정 미생물의 16S rRNA 유전자에 대한 sequencing depth가 중요하기 때문)

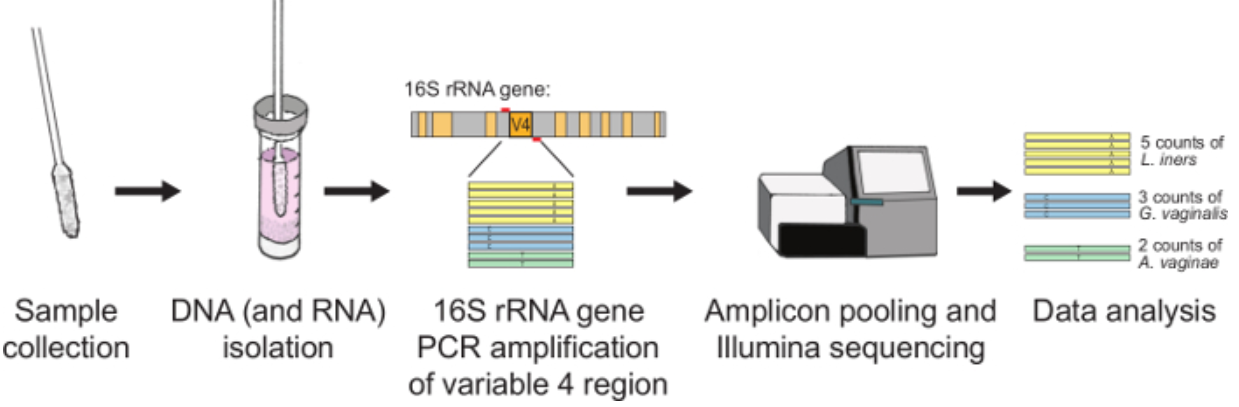
<16S rRNA 시퀀싱을 통한 마이크로바이옴 분석 과정> (그림 3, [4])
(1) 특정 환경에서 시료를 채취
(2) 시료 내 gDNA extraction
(3) 16S rRNA (V4 region)을 detection할 수 있는 primer로 PCR
(4) PCR 샘플 quality check (올바른 PCR amplicon 형성되었는지 gel loading 통한 size check 등)
(5) NGS 시퀀싱
(6) Sequence를 NCBI blast 등을 이용해 미생물 동정 (보통 Qiime이라는 플랫폼을 많이 사용, 추후 기회되면 다룰 예정)
References
[1] https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7043356/#BX1
[2] https://www.genengnews.com/commentary/sydney-brenner-a-fundamental-man/
[3] https://sueishaqlab.org/tag/16s-rrna/
[4] Bowman, Brittany A., and Douglas S. Kwon. "Efficient nucleic acid extraction and 16S rRNA gene sequencing for bacterial community characterization." JoVE (Journal of Visualized Experiments) 110 (2016): e53939.
Copyright 2020. komok’s sight All Rights Reserved.